Zawartość
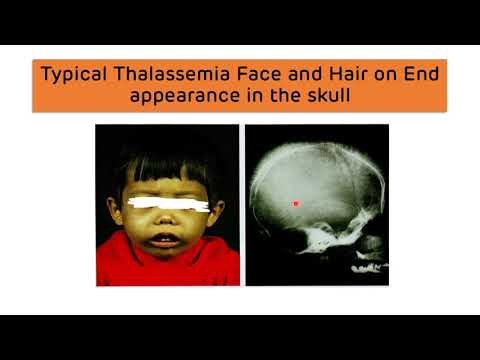
Talasemia alfa jest wrodzoną anemią, w przypadku której organizm nie jest w stanie wytworzyć normalnej ilości hemoglobiny. Hemoglobina A (główna hemoglobina u dorosłych) zawiera dwa łańcuchy alfa globiny i dwa łańcuchy beta globiny. W talasemii alfa występuje zmniejszona ilość globów alfa.Rodzaje
- Minima alfa talasemii (cichy nosiciel) występuje, gdy jeden gen alfa-globiny zostaje utracony.
- Alfa talasemia mniejsza (lub cecha) występuje, gdy utracone są dwa geny alfa globiny. Istnieją dwie formy. Kiedy dwa utracone geny alfa globiny znajdują się na tym samym chromosomie, nazywa się 16 cis, ale gdy na każdej kopii chromosomu 16 brakuje jednego genu alfa-globiny, jest to tzw przeł.
- Choroba hemoglobiny H (lub pośrednia alfa talasemia) występuje, gdy trzy geny alfa globiny nie działają. W tym przypadku występuje nadmierna ilość beta-globin. Kiedy te się łączą, nazywa się hemoglobiną H.
- Hydrops fetalis występuje, gdy utracone są wszystkie cztery geny alfa globiny. Historycznie było to niezgodne z życiem. Jeśli ryzyko jest znane z góry, transfuzje wewnątrzmaciczne (transfuzje płodu, gdy nadal jest w macicy) mogą pozwolić na udany poród. Te dzieci wymagają przewlekłej transfuzji po urodzeniu i często przeszczepu szpiku kostnego.
- Hemoglobina H Constant Spring jest odmianą talasemii alfa, w której dwa geny alfa globiny są tracone, a jeden gen alfa globiny ulega mutacji.
Kto jest zagrożony
Talasemia alfa jest powszechnie kojarzona z Azją, Afryką i obszarem śródziemnomorskim. W Stanach Zjednoczonych około 30% Afroamerykanów ma minimalne lub mniejsze wartości talasemii alfa.
Talasemia alfa jest chorobą dziedziczną i wymaga, aby oboje rodzice byli nosicielami. Osoba bez talasemii alfa powinna mieć cztery geny alfa globiny. Ryzyko urodzenia dziecka z chorobą talasemii alfa zależy od statusu rodziców. Plik przeł postać alfa talasemii mniejszej występuje częściej u osób pochodzenia afrykańskiego. Plik cis częściej występują u ludzi z Azji lub regionu śródziemnomorskiego.
Jeśli oboje rodzice mają przeł alfa talasemia minor (a- / a-), wszystkie ich dzieci będą miały przeł alfa talasemia minor. Jeśli jedno z rodziców ma cis-alfa-talasemię minor (aa / -), a drugi rodzic ma przeł alfa talasemia minor (a- / a-), mają 1 do 2 szans na urodzenie dziecka z chorobą hemoglobiny H. Podobnie, jeśli jedno z rodziców ma cis alfa talasemia minor (aa / -), a drugi rodzic ma minima talasemii (aa / a-), prawdopodobieństwo urodzenia dziecka z hemoglobiną H. wynosi 1 do 4. Hydrops fetalis występuje, gdy oboje rodzice mają cis alfa talasemia minor.
Diagnoza
Minima talasemii alfa nie powodują żadnych zmian laboratoryjnych w CBC. Dlatego nazywa się go cichym przewoźnikiem. Zwykle podejrzewa się to po urodzeniu dziecka z chorobą hemoglobiny H. Można to ustalić tylko za pomocą testów genetycznych.
Czasami alfa-talasemię mniejszą wykrywa się na ekranie noworodka, ale nie we wszystkich przypadkach. Wynik testu jest pozytywny dla hemoglobiny Bart's lub szybkich pasm. Wiele osób z mniejszą talasemią alfa nie mam pojęcia. Zwykle ujawnia się to podczas rutynowej pełnej morfologii krwi (CBC). CBC ujawni łagodną do umiarkowanej anemię z bardzo małymi czerwonymi krwinkami. Można to pomylić z niedokrwistością z niedoboru żelaza. Ogólnie rzecz biorąc, jeśli wykluczy się niedokrwistość z niedoboru żelaza i wyklucza się cechę talasemii beta, pacjent ma cechę talasemii alfa. W razie potrzeby można to potwierdzić za pomocą testów genetycznych.
Hemoglobinę H można również zidentyfikować na ekranie noworodka. Te dzieci są kierowane do hematologa w celu ścisłej obserwacji. Niektórzy pacjenci są identyfikowani w późniejszym okresie życia podczas leczenia niedokrwistości.
Hydrops fetalis nie jest specyficzną diagnozą, ale raczej charakterystyczną cechą USG noworodków. Utratę czterech genów alfa-globiny stwierdza się podczas poszukiwania przyczyny obrzęku.
Zabiegi
Nie ma potrzeby leczenia w przypadku minimów lub pomniejszych talasemii alfa. Osoby z alfa talasemią minor będą przez całe życie cierpieć na łagodną anemię.
Transfuzje: Pacjenci z hemoglobiną H mają zwykle umiarkowaną niedokrwistość, która jest dobrze tolerowana. W przypadku chorób przebiegających z gorączką czasami potrzebne są transfuje, ponieważ następuje przyspieszenie rozpadu czerwonych krwinek. Transfuzje mogą być potrzebne częściej w wieku dorosłym. Pacjenci z hemoglobiną H Constant Spring mogą mieć znaczną niedokrwistość i wymagać częstych transfuzji w ciągu życia.
Terapia chelatująca żelazo:U pacjentów z chorobą hemoglobiny H może dojść do przeciążenia żelazem nawet przy braku transfuzji krwi w wyniku zwiększonego wchłaniania żelaza w jelicie cienkim. Można je leczyć lekami zwanymi chelatami, które pomagają pozbyć się z organizmu nadmiaru żelaza.