
Zawartość
- Przyczyny
- Objawy
- Egzaminy i testy
- Leczenie
- Grupy wsparcia
- Outlook (prognozy)
- Kiedy skontaktować się z lekarzem
- Zapobieganie
- Alternatywne nazwy
- Obrazy
- Referencje
- Data przeglądu 10/15/2018
Choroba Niemanna-Picka (NPD) to grupa chorób przekazywanych przez rodziny (odziedziczone), w których substancje tłuszczowe zwane lipidami gromadzą się w komórkach śledziony, wątroby i mózgu.
Istnieją trzy powszechne formy choroby:
- Typ A
- Typ B
- Typ C
Każdy typ obejmuje różne organy. Może, ale nie musi, obejmować układ nerwowy i oddychanie. Każdy z nich może powodować różne objawy i może występować w różnym czasie przez całe życie.
Przyczyny
NPD typu A i B występują, gdy komórki w ciele nie mają enzymu zwanego kwaśną sfingomielinazą (ASM). Substancja ta pomaga w rozkładaniu (metabolizowaniu) substancji tłuszczowej zwanej sfingomieliną, która znajduje się w każdej komórce organizmu.
Jeśli brakuje ASM lub nie działa prawidłowo, sfingomielina gromadzi się wewnątrz komórek. Zabija to komórki i utrudnia prawidłowe funkcjonowanie narządów.
Typ A występuje we wszystkich rasach i grupach etnicznych. Jest to bardziej powszechne w populacji Żydów aszkenazyjskich (wschodnioeuropejskich).
Typ C występuje, gdy organizm nie może prawidłowo rozkładać cholesterolu i innych tłuszczów (lipidów). Prowadzi to do nadmiernego cholesterolu w wątrobie i śledzionie oraz do zbyt dużej ilości innych lipidów w mózgu. Typ C jest najbardziej powszechny wśród Portorykańczyków pochodzenia hiszpańskiego.
Typ C1 jest wariantem typu C. Obejmuje on defekt, który zakłóca ruch cholesterolu między komórkami mózgu. Ten typ spotykany był tylko we francuskim Kanadzie w Yarmouth County w Nowej Szkocji.
Objawy
Objawy są różne. Inne choroby mogą powodować podobne objawy. Wczesne stadia choroby mogą powodować tylko kilka objawów. Osoba może nigdy nie mieć wszystkich objawów.
Typ A zazwyczaj zaczyna się w pierwszych miesiącach życia. Objawy mogą obejmować:
- Obrzęk brzucha (obszar brzucha) w ciągu 3 do 6 miesięcy
- Wiśniowo czerwona plama z tyłu oka (na siatkówce)
- Problemy z karmieniem
- Utrata wczesnych umiejętności motorycznych (z czasem pogarsza się)
Objawy typu B są zwykle łagodniejsze. Występują w późnym dzieciństwie lub w latach nastoletnich. Obrzęk brzucha może wystąpić u małych dzieci. Nie ma prawie żadnego zaangażowania mózgu i układu nerwowego, takiego jak utrata zdolności motorycznych. Niektóre dzieci mogą mieć powtarzające się infekcje dróg oddechowych.
Typy C i C1 zazwyczaj dotyczą dzieci w wieku szkolnym. Jednak może wystąpić w dowolnym momencie pomiędzy wczesnym niemowlęctwem a dorosłością. Objawy mogą obejmować:
- Trudności z poruszaniem kończynami, które mogą prowadzić do niestabilnego chodu, niezdarności, problemów z chodzeniem
- Powiększona śledziona
- Powiększona wątroba
- Żółtaczka w (lub krótko po) porodzie
- Trudności w nauce i upadek intelektualny
- Ataki
- Niewyraźna, nieregularna mowa
- Nagła utrata napięcia mięśniowego, która może prowadzić do upadków
- Drżenie
- Problemy z poruszaniem oczami w górę iw dół
Egzaminy i testy
Test krwi lub szpiku kostnego można wykonać w celu zdiagnozowania typów A i B. Test może stwierdzić, kto ma chorobę, ale nie pokazuje, czy jest się nosicielem. Testy DNA można wykonać w celu zdiagnozowania nosicieli typów A i B.
Biopsja skóry jest zwykle wykonywana w celu zdiagnozowania typów C i D. Pracownik służby zdrowia obserwuje, jak komórki skóry rosną, poruszają się i przechowują cholesterol. Można również wykonać testy DNA w celu znalezienia 2 genów, które powodują ten typ choroby.
Inne testy mogą obejmować:
- Aspiracja szpiku kostnego
- Biopsja wątroby (zwykle niepotrzebna)
- Badanie oczu lampą szczelinową
- Testy sprawdzające poziom ASM
Leczenie
W tej chwili nie ma skutecznego leczenia dla typu A.
Przeszczepy szpiku kostnego można wypróbować dla typu B. Naukowcy nadal badają możliwe sposoby leczenia, w tym zastępowanie enzymów i terapię genową.
Nowy lek o nazwie miglustat jest dostępny dla objawów układu nerwowego typu C.
Wysoki poziom cholesterolu można kontrolować za pomocą zdrowej diety o niskiej zawartości cholesterolu lub leków. Jednak badania nie wykazują, że te metody powstrzymują nasilenie choroby lub zmieniają sposób, w jaki komórki rozkładają cholesterol. Dostępne są leki kontrolujące lub łagodzące wiele objawów, takich jak nagła utrata napięcia mięśniowego i drgawki.
Grupy wsparcia
Organizacje te mogą zapewnić wsparcie i więcej informacji na temat choroby Niemanna-Picka:
- National Institute of Neurological Disorders and Stroke - www.ninds.nih.gov/Disorders/All-Disorders/Niemann-Pick-Disease-Information-Page
- Krajowa Fundacja Chorób Niemanna-Picka - nnpdf.org
- Narodowa Organizacja ds. Rzadkich Chorób - rarediseases.org/rare-diseases/niemann-pick-disease-type-c
Outlook (prognozy)
NPD typu A jest ciężką chorobą. Zwykle prowadzi do śmierci w wieku 2 lub 3 lat.
Osoby z typem B mogą żyć do późnego dzieciństwa lub dorosłości.
Dziecko, które wykazuje objawy typu C przed 1 rokiem życia, może nie dożyć wieku szkolnego. Ci, którzy wykazują objawy po wejściu do szkoły, mogą mieszkać w wieku od średniego do późnego. Niektórzy mogą żyć do dwudziestki.
Kiedy skontaktować się z lekarzem
Umów się na spotkanie z lekarzem, jeśli masz rodzinną chorobę Niemanna-Picka i planujesz mieć dzieci. Zaleca się poradnictwo genetyczne i badania przesiewowe.
Zadzwoń do swojego usługodawcy, jeśli Twoje dziecko ma objawy tej choroby, w tym:
- Problemy rozwojowe
- Problemy z karmieniem
- Słaby przyrost masy ciała
Zapobieganie
Wszystkie typy Niemanna-Picka są autosomalne recesywne. Oznacza to, że oboje rodzice są nosicielami. Każdy rodzic ma 1 kopię nieprawidłowego genu bez żadnych oznak choroby.
Kiedy oboje rodzice są nosicielami, istnieje 25% szansy, że ich dziecko będzie chorować i 50% szansy, że ich dziecko będzie nosicielem.
Testowanie wykrywania nośnika jest możliwe tylko w przypadku zidentyfikowania wady genetycznej. Wady związane z typami A i B zostały dobrze zbadane. Dostępne są testy DNA dla tych form Niemanna-Picka.
Defekty genetyczne zostały zidentyfikowane w DNA wielu osób z typem C. Możliwe jest zdiagnozowanie osób, które niosą nieprawidłowy gen.
Kilka ośrodków oferuje testy w celu zdiagnozowania dziecka wciąż w łonie matki.
Alternatywne nazwy
NPD; Niedobór sfingomielinazy; Zaburzenia przechowywania lipidów - choroba Niemanna-Picka; Lizosomalna choroba spichrzeniowa - Niemann-Pick
Obrazy
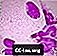
Spienione komórki Niemanna-Picka
Referencje
Kliegman RM, Stanton BF, St. Geme JW, Schor NF. Wady metabolizmu lipidów. W: Kliegman RM, Stanton BF, St. Geme JW, Schor NF, eds. Nelson Textbook of Pediatrics. 20 ed. Filadelfia, PA: Elsevier; 2016: rozdz. 86.
Patterson MC, Hendriksz CJ; NP-C Guidelines Working Group, et al. Zalecenia dotyczące diagnozy i postępowania w chorobie Niemanna-Picka typu C: aktualizacja. Mol Genet Metab. 2012; 106 (3): 330-344. PMID: 22572546 www.ncbi.nlm.nih.gov/pubmed/22572546.
Turnpenny PD, Ellard S. Wrodzone błędy metabolizmu. W: Turnpenny PD, Ellard S, eds. Emery's Elements of Medical Genetics. 15-te wyd. Filadelfia, PA: Elsevier; 2017: rozdz. 18.
Data przeglądu 10/15/2018
Zaktualizowali: Anna C. Edens Hurst, MD, MS, adiunkt w dziedzinie genetyki medycznej, University of Alabama w Birmingham, Birmingham, AL. Przegląd dostarczony przez VeriMed Healthcare Network. Recenzował także David Zieve, MD, MHA, dyrektor medyczny, Brenda Conaway, dyrektor redakcyjny i A.D.A.M. Zespół redakcyjny.